Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan V.
Tıbbi Genetik ABD Başkanı.
29.07.2025
Von Hippel-Lindau sendromu, vücudun birçok farklı yerinde tümör ve sıvı dolu keseciklerin (kistlerin) oluşumuyla karakterize kalıtsal bir hastalıktır. Tümörler kanserli veya kansersiz olabilir ve çoğunlukla genç yetişkinlik döneminde ortaya çıkar; ancak von Hippel-Lindau sendromunun belirti ve semptomları yaşam boyunca ortaya çıkabilir.
Klinik özellikler.
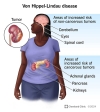
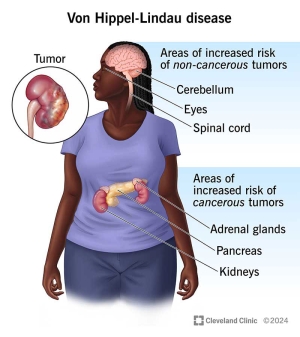
Von Hippel-Lindau sendromu (VHL), beyin, omurilik ve retinanın hemanjiyoblastomları; böbrek kistleri ve berrak hücreli renal hücreli karsinom; feokromositoma ve paraganglioma; pankreas kistleri ve nöroendokrin tümörler; endolenfatik kese tümörleri; ve epididim ve geniş ligament kistadenomları ile karakterizedir. Retina hemanjiyoblastomları VHL'nin ilk belirtisi olabilir ve görme kaybına neden olabilir. Serebellar hemanjiyoblastomlar baş ağrısı, kusma, yürüyüş bozuklukları veya ataksi ile ilişkili olabilir. Spinal hemanjiyoblastomlar ve ilgili sirinks genellikle ağrı ile ortaya çıkar. Omurilik basısı ile duyusal ve motor kayıp gelişebilir. Renal hücreli karsinom, VHL'li bireylerin yaklaşık %70'inde görülür ve önde gelen ölüm nedenidir. Feokromositomalar asemptomatik olabilir, ancak sürekli veya epizodik hipertansiyona neden olabilir. Pankreas lezyonları genellikle asemptomatik kalır ve nadiren endokrin veya ekzokrin yetmezliğe neden olur. Endolenfatik kese tümörleri, değişen şiddette işitme kaybına neden olabilir ve bu da bir başlangıç semptomu olabilir. Epididim kistadenomları nispeten yaygındır. Bilateral olmadıkları sürece nadiren sorunlara neden olurlar; bilateral ise kısırlığa yol açabilirler.
Hemanjioblastom adı verilen tümörler, von Hippel-Lindau sendromunun karakteristik özelliğidir. Bu büyümeler, yeni oluşan kan damarlarından oluşur. Genellikle kanserli olmasalar da ciddi veya yaşamı tehdit eden komplikasyonlara neden olabilirler. Beyin ve omurilikte gelişen hemanjiyoblastomlar baş ağrısı, kusma, halsizlik ve kas koordinasyon kaybına (ataksi) neden olabilir. Hemanjioblastomlar ayrıca gözün arkasını kaplayan ışığa duyarlı dokuda ( retina ) da görülebilir. Retinal anjiyomlar olarak da adlandırılan bu tümörler görme kaybına neden olabilir.
Von Hippel-Lindau sendromu olan kişilerde böbreklerde , pankreasta ve genital bölgede sıklıkla kistler gelişir. Ayrıca, berrak hücreli renal hücreli karsinom adı verilen bir böbrek kanseri türü ve pankreas nöroendokrin tümörü adı verilen bir pankreas kanseri türü geliştirme riskleri de artar.
Von Hippel-Lindau sendromu, en sık böbrek üstü bezlerinde (her böbreğin üzerinde bulunan küçük hormon üreten bezler) görülen feokromositoma adı verilen bir tümör türüyle ilişkilidir . Feokromositomalar genellikle kanserli değildir. Herhangi bir belirtiye neden olmayabilirler, ancak bazı durumlarda baş ağrısı, panik atak, aşırı terleme veya ilaçlara yanıt vermeyen tehlikeli derecede yüksek tansiyonla ilişkilidirler. Feokromositomalar, özellikle ameliyat, kaza veya hamilelik gibi stres veya travma zamanlarında tehlikelidir.
Von Hippel-Lindau sendromlu kişilerin yaklaşık %10'unda, iç kulakta kanserli olmayan tümörler olan endolenfatik kese tümörleri gelişir . Bu büyümeler, bir veya iki kulakta işitme kaybına, kulak çınlamasına (tinnitus) ve denge sorunlarına neden olabilir. Tedavi edilmezse, bu tümörler ani ve derin sağırlığa neden olabilir.
Von Hippel-Lindau sendromlu kişilerde karaciğer ve akciğerlerde kanserli olmayan tümörler de gelişebilir. Bu tümörlerin herhangi bir belirti veya semptoma neden olduğu görülmemektedir.
Von Hippel-Lindau sendromunun görülme sıklığının 36.000 kişide 1 olduğu tahmin edilmektedir.
Von Hippel-Lindau hastalığına ne sebep olur?
Von Hippel-Lindau sendromu, biyolojik ebeveynlerinizden birinin hücre büyümesini yöneten bir tümör baskılayıcı gen olan VHL geninin patojenik bir versiyonunu geçirmesiyle ortaya çıkabilen kalıtsal bir hastalıktır .
Bu genler, kansere neden olabilecek hücre büyümesini frenleyen özel proteinler üretir. Tümör baskılayıcı genler mutasyona uğradığında (değiştiğinde), frene basmaktan gaz pedalına basmaya geçerler ve hücre büyümesini aniden hızlandırırlar.
Bu hastalık otozomal dominant kalıtım örüntüsüne sahiptir. Yani biyolojik ebeveynlerinizden birinden anormal bir VHL geni miras aldıysanız, von Hippel-Lindau hastalığına yakalanma olasılığınız %50'dir. Bununla birlikte, araştırmalar VHL'li kişilerin %10'una kadarının ailesinde bu hastalığın bulunmadığını göstermektedir.
VHL genindeki mutasyonlar von Hippel-Lindau sendromuna neden olur. VHL geni, hücrelerin çok hızlı veya kontrolsüz bir şekilde büyümesini ve bölünmesini engelleyen bir tümör baskılayıcı gendir. Bu gendeki mutasyonlar, VHL proteininin üretimini engeller veya proteinin anormal bir versiyonunun üretilmesine yol açar. Değiştirilmiş veya eksik bir VHL proteini, hücrenin hayatta kalmasını ve bölünmesini etkili bir şekilde düzenleyemez. Sonuç olarak, hücreler kontrolsüz bir şekilde büyüyüp bölünerek von Hippel-Lindau sendromunun karakteristik özelliği olan tümör ve kistleri oluşturur.
VHL genindeki mutasyonlar otozomal dominant kalıtımla aktarılır ; bu, her hücrede değişmiş genin bir kopyasının tümör ve kist geliştirme riskini artırmaya yeterli olduğu anlamına gelir. Von Hippel-Lindau sendromlu çoğu kişi, etkilenen bir ebeveynden genin değişmiş bir kopyasını miras alır. Ancak vakaların yaklaşık %20'sinde, değişmiş gen, üreme hücrelerinin (yumurta veya sperm) oluşumu sırasında veya gelişimin çok erken aşamalarında meydana gelen yeni bir mutasyonun sonucudur.
Çoğu otozomal dominant durumda, her hücrede bir genin tek bir kopyasının bozukluğa neden olmak için yeterli olduğu durumlardan farklı olarak, von Hippel-Lindau sendromunda tümör ve kist oluşumunu tetiklemek için VHL geninin iki kopyasının değişmesi gerekir. VHL geninin ikinci kopyasında bir mutasyon, kişinin yaşamı boyunca beyin, retina ve böbrekler gibi organlardaki belirli hücrelerde meydana gelir. Bu genin iki kopyasının değiştiği hücreler, tümör ve kistlerin gelişmesine olanak tanıyan işlevsel VHL proteini üretmez. Bir VHL mutasyonunu miras alan hemen hemen herkes, sonunda bazı hücrelerde genin ikinci kopyasında bir mutasyon edinir ve bu da von Hippel-Lindau sendromunun özelliklerine yol açar.
Von Hippel-Lindau hastalığının tedavileri nelerdir?
Tedaviler, VHL'nin neden olduğu tümör veya kist türüne göre değişiklik gösterir. Sağlık uzmanınız tedavi seçeneklerinizi açıklayacaktır. Yaygın tedaviler şunlardır:
Tümörleri çıkarmak için yapılan ameliyat.
Kemoterapi .
Radyasyon tedavisi .
Peptit reseptör radyonüklid tedavisi ( PPRT ) ve tirozin kinaz inhibitörleri ( TKI ) dahil olmak üzere hedefli tedavi .
İmmünoterapi .
Hormon tedavisi .
VHL'nin tedavisi var mı?
von Hippel-Lindau hastalığının kesin bir tedavisi yoktur . Tedavinin amacı, tümörleri mümkün olan en erken zamanda bulup çıkarmaktır. Doktorunuz, diğer tedavilerin yanı sıra tümörleri çıkarmak için ameliyat önerebilir.
KAYNAKLAR:
https://medlineplus.gov/genetics/condition/von-hippel-lindau-syndrome/#references
http://www.ncbi.nlm.nih.gov/books/NBK1463/
https://my.clevelandclinic.org/health/diseases/6118-von-hippel-lindau-disease-vhl
Ganeshan D, Menias CO, Pickhardt PJ, Sandrasegaran K, Lubner MG, Ramalingam P, Bhalla S. Tumors in von Hippel-Lindau Syndrome: From Head to Toe-Comprehensive State-of-the-Art Review. Radiographics. 2018 May-Jun;38(3):849-866. doi: 10.1148/rg.2018170156. Epub 2018 Mar 30. Erratum In: Radiographics. 2018 May-Jun;38(3):982. doi: 10.1148/rg.2018184005.
Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145-73. doi: 10.1146/annurev.pathol.2.010506.092049. Citation on PubMed
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003 Jun 14;361(9374):2059-67. doi: 10.1016/S0140-6736(03)13643-4.
Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011 Jun;19(6):617-23. doi: 10.1038/ejhg.2010.175. Epub 2011 Mar 9. Citation on PubMed or Free article on PubMed Central
Richard S, Graff J, Lindau J, Resche F. Von Hippel-Lindau disease. Lancet. 2004 Apr 10;363(9416):1231-4. doi: 10.1016/S0140-6736(04)15957-6. No abstract available.
Sano T, Horiguchi H. Von Hippel-Lindau disease. Microsc Res Tech. 2003 Feb 1;60(2):159-64. doi: 10.1002/jemt.10253.
Shehata BM, Stockwell CA, Castellano-Sanchez AA, Setzer S, Schmotzer CL, Robinson H. Von Hippel-Lindau (VHL) disease: an update on the clinico-pathologic and genetic aspects. Adv Anat Pathol. 2008 May;15(3):165-71. doi: 10.1097/PAP.0b013e31816f852e.
Shuin T, Yamasaki I, Tamura K, Okuda H, Furihata M, Ashida S. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol. 2006 Jun;36(6):337-43. doi: 10.1093/jjco/hyl052.
van Leeuwaarde RS, Ahmad S, van Nesselrooij B, Zandee W, Giles RH. Von Hippel-Lindau Syndrome. 2000 May 17 [updated 2025 May 1]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from